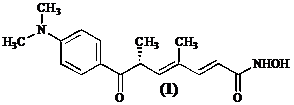
| Synthese van diketopiperazines als inhibitoren van histon deacetylase. (Gunther Roesems) |
| home | lijst scripties | inhoud | vorige | volgende |
Op wereldschaal vormen fibrotische leverziekten een belangrijk probleem in de gezondheidszorg. Leverfibrose leidt tot cirrose, en dit met de dood tot gevolg indien geen levertransplantatie wordt toegepast. In België alleen al sterven jaarlijks 1500 mensen aan fibrotische leverziekten.[1] Leverfibrose wordt niet alleen veroorzaakt door hepatitis, langdurige galstuwing en een aantal genetische leverziekten. Ook chronisch alcoholmisbruik en eiwitgebrek (bijvoorbeeld in hongergebieden) kunnen tot deze ziekte leiden.[2]
Fibrose ontstaat na een transdifferentiatie van stellaatcellen, die in normale toestand in beperkte mate bindweefsel produceren en fungeren als vitamine A reserve voor het lichaam.[1]
Fibrose en cirrose geven aanleiding tot ernstige leverstoornissen, geelzucht en oedeem. Als gevolg van de verschrompelde lever is de afvoer van het bloed uit de darmen bemoeilijkt. Hierdoor kunnen spataderen rond de slokdarm ontstaan die aanleiding kunnen geven tot een zeer grote en vaak dodelijke bloeding.[2]
Op het niveau van de celkern worden fundamentele processen zoals DNA replicatie en transcriptie geregeld door de mate waarin de e-amino groepen van de lysine residu´s der nucleosomale histonen geacetyleerd zijn. Het negatief geladen DNA is door electrostatische interactie gebonden op de histon proteïnen die rijk zijn aan positief geladen lysine residu´s. Deze residu´s worden door histon acetyl transferase geacetyleerd en door histon deacetylase kunnen de acetyl groepen verwijderd worden. Het is duidelijk dat het DNA losser gebonden is op het histon naarmate meer lysine residu´s geacetyleerd zijn (hetgeen zich voordoet wanneer het histon deacetylase wordt geïnhibeerd). Dit losser gebonden DNA leent zich beter tot expressie. Histon deacetylase inhibitoren beïnvloeden dus het fenotype van de stellaatcellen door de transcriptie van welbepaalde genen te vergemakkelijken.[1]
De actieve site van het histon deacetylase enzyme bestaat uit een hydrofobe buisvormige holte. Op de bodem bevindt zich een Zn2+ ion dat gecoordineerd is door 2 aspartaat residu´s, een histidine residu en een water molecule.[3]
Uit de bacterie Streptomyces hygroscopycus heeft men Trichostatine A (1) kunnen isoleren, een stof met goede antifibrinogene eigenschappen.[1]
Trichostatine A (1) inhibeert histon deacetylase door zijn lange alifatische keten in de hydrofobe holte te positioneren. De eindstandige hydroxamzuurfunctie bereikt aldus het polaire gedeelte van de holte waar ze met het Zn2+ ion coordineert in de hoedanigheid van een bidentaal ligand. Het ligand vervangt hierbij met zijn hydroxylgroep het aan zink gebonden watermolecule. De dimethylamino-benzoyl groep aan de andere kant van de alifatische keten maakt contacten met de ingang van de catalytische site.[3]
Gezien er geen effectieve behandeling – uitgenomen orgaantransplantatie – voor leverfibrose bestaat, spitst onderzoek zich toe op het ontwikkelen van een histon deacetylase inhibitor die de activatie van de stellaatcellen verhindert. En dit temeer omdat Trichostatine A (1) niet in voldoende mate kan verkregen worden uit Streptomyces culturen.[1] Bovendien laat de door Mori ontwikkelde asymmetrische 15 staps synthese een productie op industriele schaal niet toe.[1] en [4]
Naast Trichostatine A (1) bestaat er een familie van natuurlijk voorkomende tetracyclische peptiden, waar trapoxine B (2) en apicidine (3) deel van uitmaken, die histon deacetylase eveneens sterk inhiberen. Waar Trichostatine A (1) zijn antifibrogene werking reeds laat zien in het submicromolaire concentratiedomein (10-7 – 10-8 M), zijn de cyclische tetrapeptiden actief in het nanomolaire concentratiedomein.[1] Deze moleculen bestaan uit delen die functioneel equivalent zijn met de zinkchelator, de dimethylamino-benzoyl groep en de alifatische keten van Trichostatine A (1).[3]
In de plaats van een hydroxamzuur heeft trapoxine (2) een reactieve a-ketoepoxide functie waarop een nucleofiel uit de actieve site kan aanvallen zodat het enzyme irreversibel geïnhibeerd wordt.[3]

Apicidine (3) is het enige cyclische tetrapeptide met een ethylketon functie in de zijketen die activiteit vertoont.[1] Het ethylketon kan in interactie treden met de polaire residu´s van de actieve site, en waarschijnlijk met zink.[3]
Trapoxine (2) en apicidine (3) hebben in plaats van een dimethylamino-benzoyl groep een cyclisch tetrapeptide, met hydrofobe groepen zoals (S)-fenylalanine en (R)-proline, dat als een kap de ingang van de catalytische site kan bedekken. In vergelijking met de dimethylamino-benzoyl groep van Trichostatine A valt op te merken dat de peptidering door zijn omvang meer uitgebreide contacten kan maken met de ingang.[3]
Aangezien een diketopiperazine eenvoudiger te synthetiseren is dan een cyclisch tetrapeptide, kan men onderzoeken of cyclische dipeptiden zoals cyclo[Lys(CONHOH)-Phe] (4) een nieuwe klasse inhibitoren vormen en of ze de antifibrinogene werking van tetracyclische peptiden evenaren.
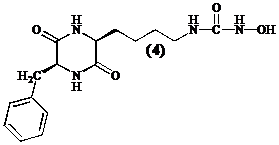
Het doel van deze thesis omvat de synthese van een nieuwe klasse histon deacetylase inhibitoren, met name diketopiperazine derivaten.
Bedenkende dat het te inhiberen enzyme als biologische functie heeft acetylaminobutyl groepen te herkennen en de verwijdering van de acetylgroepen door hydrolyse van de amide binding te katalyseren, zullen we ervoor opteren (S)-lysine te gebruiken als brug tussen de kap en de zink chelator van de inhibitor (5).
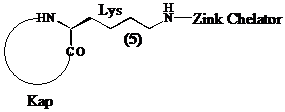
In de kapstructuur van tetracyclische inhibitoren (2 en 3) ligt tegenover de gefunctio-naliseerde alifatische zijketen met (S)-configuratie steeds een hydrofoob residu met (S)-configuratie (zoals (S)-fenylalanine bij trapoxine (2)). Daarom stellen we een diketopiperazine (6), opgebouwd uit de aminozuren (S)-lysine en (S)-fenylalanine, voor als precursor.
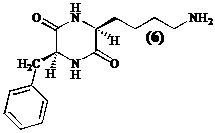
Teneinde inhibitoren te bekomen derivatiseren we de e-amino groep van lysine in functionele groepen die Zn2+ bidentaal kunnen complexeren.
Daarom wijzigen we de amino groep van de lysine zijketen als volgt:
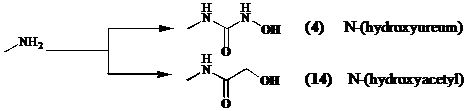
Om een derivaat te verkrijgen dat functioneel equivalent is aan het hydroxamzuur in Trichostatine A (1), synthetiseren we een N-(hydroxyureum) (4). De liganden die zorgen voor de interactie zijn de carbonyl groep van het ureum en de N-(hydroxy) groep.[15]
Bij de synthese van een tweede inhibitor, koppelt men hydroxyazijnzuur aan de lysine keten. Formeel vervangen we dus de NH in het aan lysine gelinkte hydroxamzuur door een methyleen groep. Men verwacht dat zink bidentaal gecoordineerd zal worden door de
N-(hydroxyacetyl) functie (14).[12]
5.1 Synthese van het cyclisch dipeptide
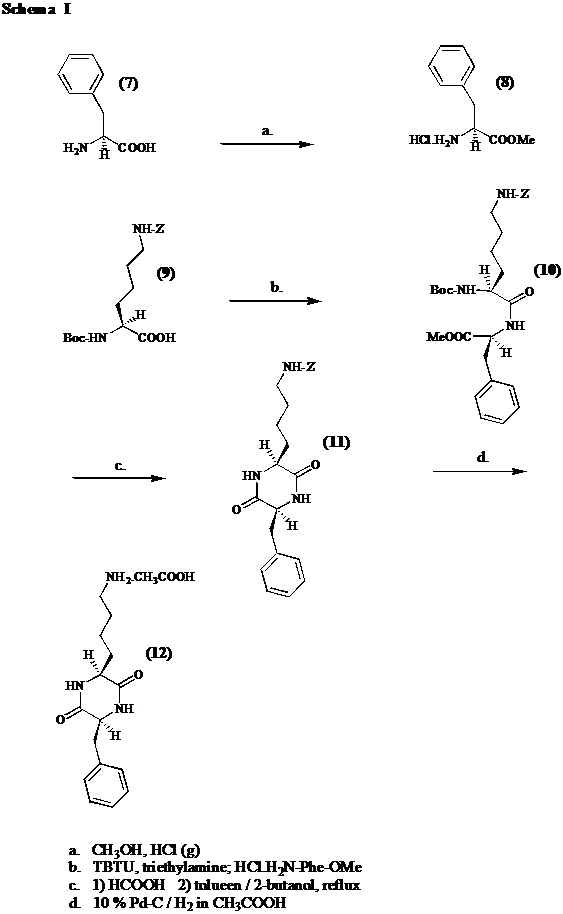
Als precursor van de verschillende histon deacetylase inhibitoren wordt cyclo[Lys-Phe] (12) gesynthetiseerd volgens schema I. In een eerste stap wordt (S)-fenylalanine (7) omgezet in zijn methyl ester (8). Vervolgens koppelt men Phe-OMe.HCl (8) aan Boc-Lys(Z)-OH (9) door het amino beschermd aminozuur (9) te activeren met TBTU. Hierna wordt de Boc beschermgroep afgesplitst door reactie met mierezuur. Het aldus ontstane dipeptide ester formaat cycliseert ter vorming van het overeenstemmende diketopiperazine (11) indien men het formaat zout refluxeert in 2-butanol / tolueen. Vooraleer tot de derivatisering over te gaan, wordt de Z groep in cyclo[Lys(Z)-Phe] (11) afgesplitst door een katalytische hydrogenolyse uit te voeren in glaciaal azijnzuur.
5.2 Derivatisering (Schema II)
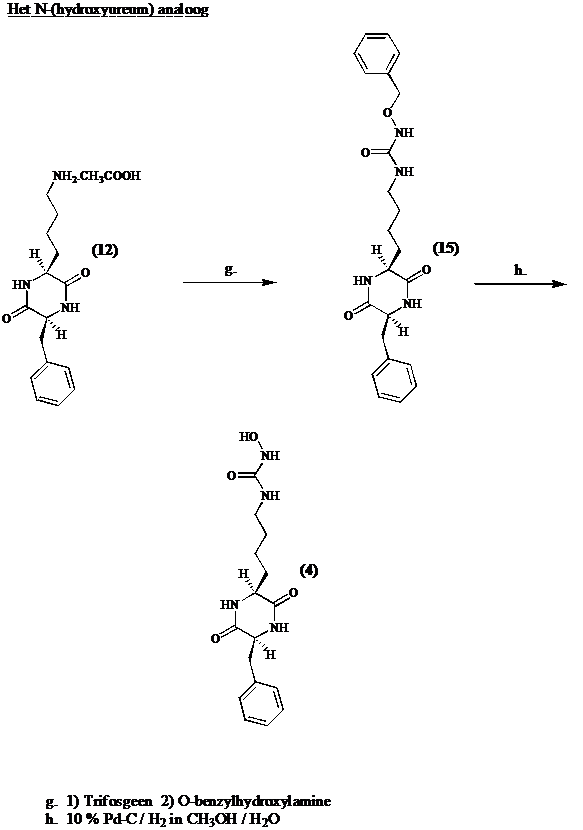
Het N-(hydroxyacetyl) analoog
De e-amino groep van de lysine zijketen wordt geacyleerd met benzyloxyacetyl chloride. Hydrogenolytische afsplitsing van de benzyl groep levert cyclo[Lys(COCH2OH)-Phe] (14).
Het N-(hydroxyureum) analoog
Men converteert cyclo[Lys-Phe] (12) in zijn isocyanaat en additie van
O-benzylhydroxylamine geeft cyclo[Lys(CONHOBzl)-Phe] (15). Na benzyl ontscherming door hydrogenolyse, bekomt men de potentiele inhibitor (4).
6.1 Synthese van (S)-fenylalanine methyl ester hydrochloride[5]
Het methyl ester van (S)-fenylalanine (8) wordt bereid door op het aminozuur (7) een Fischer verestering uit te voeren in een met HCl(g) verzadigde CH3OH oplossing. Saturatie van de oplossing dient wegens de exothermiciteit te gebeuren in een ijsbad. De reactie zelf verloopt 4 uur bij kamertemperatuur. De eindopbrengst bedraagt 72 % na kristallisatie.
6.2 Synthese van Boc-Lys(Z)-Phe-OMe[6]
Men koppelt Phe-OMe.HCl (8) aan Boc-Lys(Z)-OH (9) in de aanwezigheid van3 eq. triethylamine door van het laatstvernoemde aminozuur (9) een actief ester te maken met 1.1 eq. TBTU. 1 eq. triethylamine wordt verbruikt om Boc-Lys(Z)-OH (9) om te zetten in zijn carboxylaat ion, wat vervolgens met TBTU reageert. Men merkt op dat er zich nog 0.1 eq. TBTU in het reactiemidden bevindt. Indien men TBTU in overmaat gebruikt, moet men ook Phe-OMe.HCl (8) in overmaat gebruiken. Men weet immers dat TBTU reageert met aminogroepen ter vorming van een Schiff base. 0.1 eq. Phe-OMe.HCl (8) kan deze nevenreactie ondergaan, terwijl 1 eq. moet overblijven om met het actieve ester te reageren. Daarom wordt 1.1 eq. Phe-OMe.HCl (8) toegevoegd, na een activatieperiode van 10 minuten. 1.1 eq. triethylamine wordt verbruikt om het amine (8) uit zijn zout vrij te zetten. Men bekomt na kristallisatie een opbrengst van 88 %.
Algemeen geldt voor TBTU koppelingen dat er even veel of minder racemisatie optreedt dan met carbodiimide. HPLC wijst niet uit dat racemisatie door oxazolonvorming zou hebben plaatsgevonden.
6.3 Synthese van cyclo[Lys(Z)-Phe][7]
Om van Boc-Lys(Z)-Phe-OMe (10) een diketopiperazine (11) te vormen, splitst men in een eerste stap de Boc groep af in mierezuur. Bij het evaporeren van het mierezuur dient men er vooral op te letten dat het waterbad niet warmer wordt dan 30 °C. Vervolgens refluxeert men het aldus ontstane dipeptide formaat zout in een 3 / 1 mengsel van 2-butanol / tolueen bij
98 °C. Tijdens het refluxeren wordt het mierezuur als een azeotroop verwijderd, waardoor het dipeptide achterblijft om te cycliseren.[8] Door de lage diëlectrische constante van het solventsysteem wordt significante epimerisatie voorkomen.[9] Gezien de hoge diëlectrische constante van mierezuur (58.5), mag men de temperatuur niet laten oplopen bij het uitdampen. Na cyclo[Lys(Z)-Phe] (11) te laten neerslaan, bedraagt de netto opbrengst 85 %. Er waren geen aanwijzingen in het HPLC chromatogram en op de dunne laag dat tijdens de ringvorming epimerisatie zou hebben plaatsgevonden.
6.4 Synthese van cyclo[Lys-Phe][10]
Men splitst de Z groep af in cyclo[Lys(Z)-Phe] (11) door een katalytische hydrogenolyse uit te voeren in glaciaal azijnzuur. Men gebruikt 0.5 gewichtsequivalent 10 % Pd / C als heterogene katalysator. De hydrogenolyse gebeurt bij 60 psi gedurende 2 uur. Uiteindelijk bekomt men een opbrengst van 94 %. Uit het HPLC chromatogram blijkt echter dat 4 % geëpimeriseerd is. Door 2 omkristallisaties hebben we getracht het percentage epimerisatie te reduceren. De meetfout in achting nemende, kon worden opgetekend dat geen significante verbetering van de zuiverheid werd waargenomen. Om ons ervan te verzekeren dat de epimerisatie niet is opgetreden wegens de zure condities bij de hydrogenolyse, lossen we de kristallen cyclo[Lys-Phe] (12) op in glaciaal azijnzuur. Na 8 uur is geen verdere epimerisatie opgetreden. Vermits in oplossingen met hoge diëlectische constante reeds bij 70 °C epimerisatie kan optreden,[9] werd nagegaan of epimerisatie niet is opgetreden bij de zuivering van cyclo[Lys(Z)-Phe] (11) waar hete ethylacetaat werd gebruikt. Hiertoe werd opnieuw cyclo[Lys(Z)-Phe] (11) gesynthetiseerd en zonder voorafgaande zuivering werd het aan een hydrogenolyse onderworpen. In het chromatogram verscheen de epimeerpiek. Epimerisatie treedt dus niet op tijdens de zuivering. De chirale zuiverheid werd niet bewaard bij de koppeling of bij de cyclisatie.
6.5 Synthese van cylco[Lys(COCH2OBzl)-Phe][11]
De reactie van cyclo[Lys-Phe] (12) met benzyloxyacetylchloride wordt onder de getabelleerde condities uitgevoerd:
|
Experiment |
Solvent |
Base |
# eq. zuurchloride |
Reactieduur |
Opbrengst |
|
1[12] |
CH2Cl2 |
5 eq. pyridine |
2 eq. toegevoegd bij 0°C |
28 u bij kt |
34 % bruto (1 piek op HPLC) |
|
2[11] |
CH2Cl2 |
2.4 eq. NaOH |
1.3 eq. toegevoegd bij kt |
6 u 30 bij kt |
69 % kristallen |
Er wordt in experiment 1 een overmaat van 2 eq. pyridine gebruikt omdat deze base assisteert bij de nucleofiele acyl substitutie door reactie met het zuurchloride. Hierbij wordt een acyl pyridinium intermediair gevormd.[13] Men ziet op HPLC dat zelfs na 28 uur geen verdere conversie is opgetreden. We halen de slechte oplosbaarheid van het diketopiperazine (12) aan als oorzaak van de lage opbrengst.
Omdat cyclo[Lys-Phe] (12) oplost in een heftig geroerd CH2Cl2 / NaOH (aq) systeem, schakelen we over op de Schotten Baumann procedure.[11] Hierbij wordt gewoonlijk een kleine overmaat (8 tot 15 %) NaOH gebruikt.[14]. Wegens oplosbaarheidsproblemen ziet men ervan af het zuurchloride bij 0 °C toe te voegen.
Men ziet duidelijk dat het aanwenden van Schotten Baumann condities bij deze synthese een vereiste is.
6.6 Synthese van cylco[Lys(COCH2OH)-Phe][12]
Door een katalytische hydrogenatie uit te voeren op cyclo[Lys(COCH2OBzl)-Phe] (13), splitst men de benzyl groep af. Als solventsysteem gebruikt men een 6 / 1 mengsel van methanol / water en 0.5 gewichtsequivalent 10 % Pd/C fungeert als katalysator. De hydrogenolyse verloopt bij 48 psi. Na 5 u 30 ziet men op het chromatogram dat de benzyl ontscherming slechts voor 92 % is doorgegaan. Daarom laat men de reactie nog 3 uur verder verlopen. HPLC wijst nu uit dat de conversie volledig is opgetreden. Rekristallisatie uit warme ethylacetaat levert een opbrengst van 95 %.
We merken op dat bij een vergelijkbare reactie in de literatuur mildere condities volstaan (0.17 g ewichtsequivalent 10 % Pd/C, hydrogenolyse bij 15 psi gedurende 6 uur).[12]
6.7 Synthese van cylco[Lys(CONHOBzl)-Phe]
Verschillende synthese strategieën werden onderzocht om de meest gunstige condities te vinden voor de bereiding van cyclo[Lys(CONHOBzl)-Phe] (15).
6.7.1 Reacties met trifosgeen
Onderstaande tabel laat zien hoe men met trifosgeen een amine in zijn isocyanaat omzet. Dit isocyanaat laat men vervolgens reageren met een tweede amine om een ureum te bekomen.
|
|
Vorming van het isocyanaat |
Vorming van het ureum |
||||
|
Experiment |
Amine |
# eq. trifosgeen |
Reactieduur |
Voeg isocyanaat bij |
Reactieduur |
Opbrengst |
|
1[15] |
cyclo[Lys-Phe] |
0.37 eq. |
1 u |
2 eq. O-benzyl- hydroxylamine |
2 u |
9 % (22 % bruto) |
|
2 |
cyclo[Lys-Phe] |
0.7 eq. |
1u |
2 eq. O-benzyl- hydroxylamine |
2 u |
24 % (60 % bruto) |
|
3 |
O-benzyl- hydroxylamine |
0.33 eq. |
1 u 30 |
0.5 eq. cyclo[Lys-Phe] |
2 u 30 |
0 % |
|
4 |
O-benzyl- hydroxylamine |
0.33 eq. |
30 minuten |
0.5 eq. n-propylamine |
2 u |
0 % |
|
5 |
n-propylamine |
0.37 eq. |
40 minuten bij 0 °C |
2 eq O-benzyl- hydroxylamine |
2 u bij 0 °C |
0 % |
Algemene Opmerking: Bij testreacties gebruikt men n-propylamine in plaats van cyclo[Lys-Phe]¨(12). Zo beperkt men het verbruik van het te derivatiseren diketopiperazine.
Bij de vorming van het isocyanaat, die optreedt in op in een CH2Cl2 / H2O bifasisch systeem dat 3 eq Na2CO3 bevat, wordt een gelachtige pap gevormd. Er treedt mechanisch verlies op terwijl men deze pap bij het hydroxylamine voegt.
In experiment 1 bekomt men een bruto opbrengst van 22 %. HPLC wijst uit dat de bruto opbrengst voor 43 % bestaat uit cyclo[Lys(CONHOBzl)-Phe] (15). Door kristallisatie werd bijgevolg alle eindproduct (15) afgezonderd. Naast de piek van het eindproduct, ziet men op het chromatogram van de bruto opbrengst een ruis van een 15 tal ongeïdentificeerde nevenproducten. Men vindt het ongereageerde cyclo[Lys-Phe] (12) tesamen met
O-benzylhydroxylamine terug in de HCl fase. Dit wijst op dat een onvolledige reactie.
In de literatuur bekomt men voor een vergelijkbare reactie een opbrengst van 48 %.[15]
Bij experiment 2 ziet men dat de opbrengst substantieel stijgt als men de overmaat trifosgeen vergroot. We vertrouwen erop dat door kristallisatie alle eindproduct werd afgezonderd.
We schrijven de lage opbrengst toe aan de slechte oplosbaarheid van cyclo[Lys-Phe].
Omdat het diketopiperazine (12) oplosbaar is in een basisch CH3CN / H2O mengsel onderzoeken we in experiment 5 of de reactie niet uitvoerbaar is in zulk een homogeen midden. Men stelt echter in deze testreactie met n-propylamine vast dat uitsluitend
N,N’-dipropylureum wordt gevormd. Aldus is de poging om het oplosbaarheidsprobleem te omzeilen, mislukt.
Het zou interessant zijn om O-benzylhydroxylamine in zijn isocyanaat om te zetten omdat men zo een grote overmaat isocyanaat kan toevoegen aan het diketopiperazine (12). Men neemt waar dat O-benzylhydroxylamine met zijn eigen isocyanaat reageert ter vorming van
volgend symmetrisch ureum (16):
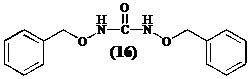
Cyclo[Lys-Phe] (12) wordt integraal teruggevonden in de waterfase. In een volgend experiment heeft men getracht de vorming van N,N’-di(O-benzylhydroxy)ureum (16) te vermijden door:
1) Het hydroxylamine langzaam toe te druppelen aan de trifosgeenoplossing zodat er steeds een overmaat trifosgeen aanwezig is.
2) De reactietijd voor de vorming van het isocyanaat terug te brengen tot 30 minuten.
De vorming van het asymmetrisch ureum treedt niet op, alleen (16) wordt teruggevonden..
6.7.2 Reactie met p-nitrofenyl chloroformaat
We onderzoeken in de volgende testreacties of het niet mogelijk is de opbrengst te verhogen met p-nitrofenyl chloroformaat.
Men laat n-propylamine reageren met p-nitrofenyl chloroformaat tot het overeenkomstige carbamaat, dat vervolgens wordt toegevoegd aan O-benzylhydroxylamine om zo het ureum te vormen.
|
Vorming van het carbamaat |
Vorming van het ureum |
|||||
|
Experiment |
Amine |
Voeg amine bij |
Reactieduur |
Voeg carbamaat bij |
Reactieduur |
Opbrengst |
|
1[16] |
n-propyamine + 1 eq. DiPEA |
CH2Cl2 + 1 eq. p-nitrofenyl chloroformaat |
1 u bij 0 °C |
1.5 eq. O-benzyl- hydroxylamine + 4 eq. DiPEA |
3 u bij kt |
8 % |
|
2[17] |
n-propylamine + 1.2 eq. DiPEA |
CH2Cl2 + 1.2 eq. p-nitrofenyl chloroformaat + 0.1 eq. DMAP |
1 u bij 0 °C |
2 eq. O-benzyl- hydroxylamine + 4 eq. DiPEA |
3 u bij kt |
17 % |
Opmerking: Men schat de opbrengst op basis van een HPLC analyse op de bruto opbrengst.
Bij de vorming van het carbamaat is het van belang n-propylamine druppelsgewijs toe te voegen aan de p-nitrofenyl chloroformaat oplossing. Men wil immers vermijden dat
n-propylamine met het gevormde carbamaat reageert ter vorming van N,N’-dipropylureum. In experiment 1 kleurt het reactiemidden geel na 35 minuten (p-nitrofenol). Dit wijst op het optreden van de hoger geschetste nevenreactie. Men heeft niet kunnen quantificeren hoeveel nevenproduct gevormd werd gezien bij de afwerking veelvuldig gewassen moet worden met een K2CO3 (aq) oplossing om p-nitrofenol te extraheren. Hierbij migreert het symmetrisch ureum naar de waterfase.
Men heeft de opbrengst kunnen verhogen door:
1) Een overmaat p-nitrofenyl chloroformaat te gebruiken
2) DMAP te gebruiken om de vorming van het carbamaat te versnellen.
Nu kleurt het reactiemidden na 50 minuten geel.
We besluiten dat deze strategie geenszins tot verbetering leidt vermits:
1) De opbrengst lager is
2) Symmetrisch ureum als nevenproduct gevormd wordt
3) De afwerking langer is.
6.7.3 Reactie met fenyloxycarbonylsuccinimide[18]
Om de opbrengst te verbeteren stellen we voor het meer stabiele fenyl carbamaat te synthetiseren. Hierdoor zal de vorming van het symmetrisch ureum voorkomen worden. Het fenyl carbamaat zal men afzonderen en onderwerpen aan een reactie met
O-(t-butyldimethylsilyl)hydroxylamine.
Vorming van het fenyl carbamaat
In een 4 /2 oplossing van dioxaan / water laat men n-propylamine gedurende 1 uur reageren met 1 eq. fenyloxycarbonylsuccinimide. De bruto opbrengst is zuiver en bedraagt 96 %.
Vorming van het ureum
We proberen een reactie teweeg te brengen tussen het hierboven gevormde carbamaat en 1eq.
O-(t-butyldimethylsilyl)hydroxylamine onder de volgende condities:
|
Solvent |
Temperatuur |
Reactieduur |
Bevinding |
|
dioxaan |
50 °C |
1 dag |
geen reactie |
|
dioxaan + 1 eq. DiPEA |
80 °C |
2 dagen |
geen reactie |
Het reactieverloop werd gevolgd met HPLC.
De conclusie luidt dat het carbamaat te weinig reactief is.
6.8 Synthese van cyclo[Lys(CONHOH)-Phe]
We stellen voor de benzyl ontscherming op cyclo[Lys(CONHOBzl)-Phe] (15) te laten verlopen onder dezelfde omstandigheden als in § 6.6 omdat de hydrogenolyse op cyclo[Lys(COCH2OBzl)-Phe] (13) slechts verliep onder condities die – in vergelijking met deze uit de literatuur- meer drastisch zijn.
Na kristallisatie bekomt men slechts een opbrengst van 68 %.
Een aantal bevindingen suggereren dat tijdens de hydrogenolyse het hydroxyureum partieel gereduceerd werd tot cyclo[Lys(CONH2)-Phe].
Op het chromatogram vindt men dat de hoofdpiek (Rt = 10.69) niet is opgesplitst. Verder treft men een onzuiverheid aan (Rt = 14.5; 7.4 %).
LC /MS bevestigt dat de hoofdpiek niet is opgesplitst en geeft voor deze piek de volgende fragmentatie:
LC / MS (ES+):
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
302 |
M – NHOH of M’ – NH2 |
100 % |
|
319 |
M – O + H of M’ + H |
80 % |
|
335 |
M + H |
55 % |
met M = cyclo[Lys(CONHOH)-Phe]
M’ = cyclo[Lys(CONH2)-Phe]
Er elueert verder geen piek met m/z = 319.
Massaspectrometrie laat zien dat er niet alleen een M + Na piek, maar ook een M’ + Na piek wordt aangetroffen:
MS (ES+):
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
302 |
M – NHOH of M’ – NH2 |
35 % |
|
319 |
M’ + H |
35 % |
|
335 |
M + H |
35 % |
|
341 |
M’ + Na |
100 % |
|
357 |
M + Na |
40 % |
Dit doet vermoeden dat de piek met m/z = 319 staat voor M’ + H.
Het nevenproduct zou in dit geval dezelfde retentietijd hebben als het hydroxyureum.
We onderzoeken de situatie verder met 1H NMR.
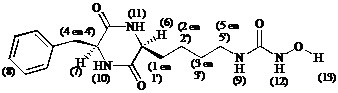
NMR: (500 MHz; spectrumopname bij 303 K met presaturatie)
Solvent: CDCl3 / dimethyl-d6 sulfoxide (3 / 1)
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
Koppelingsconstante J in Hz. |
|
CH(1’) |
0.7 |
1 H |
m |
|
|
CH(2, 2’) |
0.9 tot 1.0 |
2 H |
M |
|
|
CH(1, 3, 3’) |
1.1 tot 1.3 |
3 H |
M |
|
|
CH(4’, 5, 5’) |
2.9 tot 3.0 |
3 H |
M |
|
|
CH(4) |
3.1 |
1 H |
dd |
Jgem = 13.6 en Jvic = 5.1 |
|
CH(6) |
3.6 |
1 H |
m |
|
|
CH(7) |
4.1 |
1 H |
m |
|
|
Ph(8) |
7.1 tot 7.2 |
5 H |
M |
|
|
NH(9) |
|
|
|
|
|
NH(10) |
7.65 |
1 H |
s |
|
|
NH(11) |
7.75 |
1 H |
s |
|
|
NH(12) |
|
|
|
|
|
OH(13) |
|
|
|
|
De ureum protonen (9) en(12) die men respectievelijk verwacht bij d » 6.5 ppm en d » 9 ppm, vindt men niet terug. Het singlet bij d = 7.62 ppm geeft geen kruispieken in het COSY spectrum. We veronderstellen derhalve dat dit signaal afkomstig is van de cyclo[Lys(CONH2)-Phe] protonen.
Uit feit dat het hydroxylproton (13) niet wordt teruggevonden, kan men geenszins besluiten dat er reductie heeft plaatsgevonden. Het signaal kan door presaturatie of uitwisseling zijn onderdrukt.
Alles in beschouwing nemend, vermoeden we dat een afsplitsing van de hydroxyl groep als nevenreactie is opgetreden.
Het doel van deze thesis was de synthese van diketopiperazine derivaten die zouden kunnen fungeren als histon deacetylase inhibitoren.
Als precursor diende cyclo[Lys-Phe] (12) bereid te worden.
Men vertrekt hiervoor van (S)-Phe (7). Fischer verestering leverde het methyl ester (8) met een opbrengst van 72 %. Na Boc-Lys(Z)-OH (9) te hebben geactiveerd met TBTU, koppelde men daaraan Phe-OMe.HCl (8). Men verkreeg Boc-Lys(Z)-Phe-OMe (10) met een goede opbrengst (88 %). Om een diketopiperazine te vormen, splitste men de Boc groep af in mierezuur waarna het dipeptide formaat zout onder reflux cycliseerde tot cyclo[Lys(Z)-Phe] (11) (85 %). Na de Z ontscherming (94 %), bleek dat tijdens de koppeling of tijdens de ringvorming de chirale zuiverheid niet behouden bleef. Er trad 4 % epimerisatie op.
Om het N-(hydroxyacetyl) analoog (14) te bekomen, werd onder Schotten Baumann condities cyclo[Lys-Phe] (12) geacyleerd met benzyloxyacetylchloride (69 %). Benzyl ontscherming op cyclo[Lys(COCH2OBzl)-Phe] (13) verliep zonder problemen (95 %).
Inzake het N-(hydroxyureum) (4) hebben we aangetoond dat het aangewezen was cyclo[Lys(CONHOBzl)-Phe] (15) te bereiden via de trifosgeen route. Met p-nitrofenyl chloroformaat trad vorming van een symmetrisch ureum op als nevenreactie, en daarenboven was de opbrengst lager en verliep de extractie van p-nitrofenol moeilijk. Reactie met fenyloxycarbonylsuccinimide leverde ons een carbamaat met een uitstekende opbrengst. Het probeem verlegde zich hier echter naar de vorming van het ureum.
Toen de benzyl ontscherming op cyclo[Lys(CONHOBzl)-Phe] (15) werd doorgevoerd, trad afsplitsing van de hydroxylgroep op als nevenreactie
8.1 Identificatietechnieken en zuiverheidsbepaling
8.1.1 Smeltdomein
Het smeltdomein wordt bepaald met een Buchi B-540 toestel. Men stelt het temperatuursincrement in op 1 °C / min.
8.1.2 Optische rotatie
Men meet de optische rotatie a met een Optical Activity AA-5 polarimeter. De specifieke rotatie wordt berekend met:
[a]D = a / c . l
waarbij c = de concentratie in g / ml.
l = de lengte van de meetcel in dm.
8.1.3 Thin Layer Chromatography (TLC)
Er wordt gebruik gemaakt van met silica bedekte glasplaatjes (Merck 60, lengte 10 cm). In de silicagel heeft men een fluorescentie indicator (F254) verwerkt om de spots na een I2 test of onder UV licht te kunnen visualiseren. Producten met amide bindingen kan men detecteren na een toluidine test. Voor primaire aminen gebruikt men de ninhydrine test.
8.1.4 High Pressure Liquid Chromatography (HPLC)
De chromatogrammen worden opgenomen door het Spectra Physics P4000 toestel.
De reversed phase kolom is van het type Vydac 218 TP 54. De Spectrasystem UV 2000 detector stelt men in op 215 nm.
Men maakt gebruik van de volgende elutiegradient:
|
Tijd / min |
Debiet in ml / min |
Solvent A H2O + 0.1 % TFA |
Solvent B 8 / 2 CH3CN / H2O + 0.1 % TFA |
|
0 |
1 |
100 % |
0 % |
|
30 |
1 |
0 % |
100 % |
|
35 |
1 |
0 % |
100 % |
8.1.5 Massaspectrometrie (MS)
De massaspectra worden opgenomen met een VG Quattro II spectrometer. Ionisatie wordt door electrospray teweeggebracht. De intensiteiten van de belangrijkste pieken worden relatief t.o.v. de intensiteit van de hoofdpiek weergegeven. Tenzij anders vermeld, wordt acetonitril gebruikt bij de staalvoorbereiding.
8.1.6 Nuclear Magnetic Resonance Spectroscopy (NMR)
Na elke reactiestap wordt een 1H NMR spectrum opgenomen met een Bruker AC-250
(250 MHz) spectrometer. De gebruikte solventen worden expliciet vermeld.
Voor de karakterisatie van het eindproduct (14) wordt gebruik gemaakt van een 500 MHz spectrometer. Spectrumopname vindt plaats bij 303 K met presaturatie. De interpretatie van het hiermee bekomen 1D spectrum werd uitgevoerd met behulp van 2D COSY en 2D TOCSY (mengtijd = 57 ms).
Met het nauwkeurig geïnterpreteerde spectrum van (14) in handen, werden de signalen in de 250 MHz spectra toegekend via een teruggaande analyse. We hebben hiervoor ook gekeken naar de random coil chemical shifts van de aminozuren Lys en Phe.[21]
Inzake de nomenclatuur spreken we af dat het diastereotope proton dat voorkomt bij hoog veld wordt gespecificeerd met een accent.
8.1.7 Infrarood spectroscopie (IR)
Men neemt het IR spectrum op met een Perkin-Elmer 257 toestel. Als staalvoorbereiding perst men een pastille die 1 mg vast product en 70 mg KBr bevat.
8.2 Synthese van cyclo[Lys-Phe] (12)
8.2.1 Karakterisatie van (S)-fenylalanine (7)

Witte vaste stof.
Moleculair gewicht: 165.19 g/mol
Brutoformule: C9H11NO2
Ter controle van de zuiverheid verifiëren we dat het aminozuur (7) begint te ontbinden bij
274 °C.[19]
We nemen degradatie van (S)-fenylalanine (7) waar bij 273 °C.
8.2.2 Synthese van (S)-fenylalanine methyl ester hydrochloride (8)[5]
In een 250 ml eenhalzige kolf, voorzien van een gasinlaat en een lege CaCl2 droogbuis die fungeert als gasuitlaat, voegt men 150 ml methanol bij 10.08 g (61 mmol) (S)-fenylalanine (7). Het reactiemidden wordt magnetisch geroerd terwijl HCl(g) door het midden geborreld wordt. Men stopt wanneer alle (S)-fenylalanine (7) is opgelost. Men koelt de kolf met een ijsbad en satureert de oplossing met HCl(g). Verzadiging wordt bereikt als een nat pH-papiertje rood kleurt aan de gasuitlaat. We laten de oplossing 4 uur staan bij kamertemperatuur na een CaCl2 droogbuis op de kolf te hebben gezet om de oplossing te beschermen tegen de luchtvochtigheid. Men merkt op dat het gevormde zout (8) oplosbaar is in methanol. Men evaporeert het solvent, erop toeziend dat de temperatuur van het waterbad de drempel van 50 °C niet overschrijdt. We lossen het residu op in 50 ml methanol en herhalen de uitdamping. Vervolgens wordt het droge residu gewassen met 75 ml diethylether. De verkregen neerslag wordt gefilteerd op een glasfilter en gedroogd onder vacuüm over NaOH pellets om de overmaat HCl te neutraliseren. De bruto opbrengst bedraagt 98 % (12.94 g; 60 mmol). Ter zuivering rekristalliseren we het ester hydrochloride (8) uit 50 ml methanol door geleidelijke toevoeging van 200 ml diethylether. Nadat uitvlokking wordt waargenomen, zet men de erlenmeyer 4 uur in de koelkast. De kristallen giet men af op een glasfilter, waarna ze in een dessicator worden gedroogd. Vermits de opbrengst slecht 39 % (5.25 g; 24 mmol) bedraagt, dampt men het filtraat uit. Dit doen we in de verwachting bijkomend product uit het residu te isoleren. Het residu wordt gewassen met 75 ml diethylether en de suspensie giet men af op een glasfilter. Rekristallisatie vindt plaats onder hoger vermelde condities. De na filtratie verkregen kristallen worden gedroogd en verhogen de netto opbrengst met 2.79 g. Deze recuperatieprocedure wordt een laatste keer met succes op het filtraat toegepast en levert ons 1.53 g product, hetgeen de totale opbrengst brengt op 72 % (9.57 g; 44 mmol)
Karakterisatie:

Witte vaste stof.
Moleculair gewicht: 215.68 g/mol
Brutoformule: C10H14ClNO2
Smeltdomein: 155.9 - 157 °C
Literatuurwaarde: 158 – 160 °C[20]
Optische rotatie:
Solvent: ethanol
Concentratie c = 0.0103 g/ml
[a]D= +36 °
Literatuurwaarde: [a]D = +38.0 ± 1 voor c = 0.002 g/ml in ethanol. Zuiverheid > 99%[20]
TLC:
Eluens 1: dichloormethaan
Rf = 0
Eluens 2: ethylacetaat / methanol (4 / 1)
Rf = 0.44
Eluens 3: acetonitril / methanol / water (4 / 1 / 1)
Rf = 0.61
HPLC:
|
Retentietijd/min |
Toekenning |
|
11.3 |
(S)-Phe-OMe.HCl |
MS (ES+):
De molaire massa van het methyl ester bedraagt 179 g/mol.
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
120 |
M – COOCH3 |
100 % |
|
180 |
M + H |
75 % |
|
359 |
2M + H |
5 % |
NMR: (250 MHz)
Solvent: dimethyl-d6 sulfoxide
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
Koppelingsconstante J in Hz. |
|
CH(1) |
3.22 |
1 H |
dd |
Jgem = 14.0 en Jvic = 5.3 |
|
CH(1’) |
3.09 |
1 H |
dd |
Jgem = 13.9 en Jvic = 7.5 |
|
CH3(2) |
3.63 |
3 H |
s |
|
|
CH(3) |
4.18 |
1 H |
m |
|
|
Ph(4) |
7.15 tot 7.36 |
5 H |
M |
|
|
NH+3(5) |
8.68 tot 8.91 |
3 H |
bs |
|
IR:
n (in cm-1): 3230 tot 2400; 1745; 1590 en 1500; 1300; 1240; 750 en 710
8.2.3 Synthese van Boc-Lys(Z)-Phe-OMe (10)[6]
In een droge 250 ml kolf lost men onder magnetisch roeren 3 g (7.89 mmol) Boc-Lys(Z)-OH (9) op in 35 ml droge DMF. De oplossing wordt basisch gemaakt met 3 eq (3.29 ml; 23.67 mmol) triethylamine. Vervolgens voegt men 1.1 eq TBTU (2.79g; 8.68 mmol) toe. Na 10 minuten voegt men bij geactiveerde lysine 1.1 eq Phe-OMe.HCl (8) (1.87g; 2.68 mmol). Men giet een additionele hoeveelheid van 25 ml DMF in het reactiemidden. Het reactieverloop wordt met HPLC gevolgd. Na 1 uur blijkt dat de oppervlakte van de Boc-Lys(Z) piek ten opzichte van de dipeptidepiek niet meer afneemt. De verhouding der oppervlaktes van deze pieken bedraagt 0.04 (= Boc-Lys(Z) / dipeptide). Men dampt na 2 uur het solvent uit met een oliepomp. Hierbij verwarmt men de kolf tot 50 °C. De olie lost men op in 80 ml ethylacetaat en men wast sequentieel de organische fase
3 keer met een 25 ml 2N citroenzuuroplossing,
3 keer met een 25 ml 5% ige NaHCO3 oplossing,
3 keer met een 25 ml 5% ige NaCl oplossing.
De organische fase wordt afgezonderd en gedroogd over MgSO4. Nadien wordt het MgSO4 gespoeld met 70 ml ethylacetaat. Het filtraat wordt uitgedampt en de bruto opbrengst bedraagt 6.61 g (100 %). De verkregen olie lost men op in 30 ml diethylether, door geleidelijke toevoeging van 50 ml petroleumether kristalliseert het dipeptide (10) uit. De witte kristallen filtreert men af op een glasfilter, waarna ze gewassen worden met 50 ml petroleumether. De opbrengst na drogen onder vacuum bedraagt 88 % (3.77 g; 6.96 mmol).
Karakterisatie:

Witte vaste stof.
Moleculair gewicht: 541.64 g/mol
Brutoformule: C29H39N3O7
Smeltdomein: 101.3 – 103.5 °C
Optische rotatie:
Solvent: CH2Cl2
Concentratie c = 0.0102 g/ml
[a]D= +18 °
TLC:
Eluens 1: hexaan / ethylacetaat (4 / 1)
Rf = 0
Eluens 2: ethylacetaat / petroleumether (7 / 3)
Rf = 0.47
Eluens 3: ethylacetaat / methanol (4 / 1)
Rf = 0.73
De dipeptide vlek werd enkel zichtbaar na een toluidine test.
HPLC:
|
Retentietijd/min |
Toekenning |
|
25.9 |
Boc-Lys(Z)-Phe-OMe |
MS (ES+):
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
442 |
M + H – Boc |
20 % |
|
542 |
M + H |
60 % |
|
564 |
M + Na |
100 % |
|
580 |
M + K |
75 % |
NMR: (250 MHz)
Solvent: CDCl3
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
Koppelingsconstante J in Hz. |
|
CH(1, 1’, 2, 2’, 3, 3’) |
1.1 tot 1.7 |
6 H |
M |
|
|
CH3(4) |
1.3 |
9 H |
s |
|
|
CH(5, 5’, 6, 6’) |
2.9 tot 3.1 |
4 H |
M |
|
|
CH3(7) |
3.6 |
3 H |
s |
|
|
CH(8) |
3.9 tot 4 |
1 H |
M |
|
|
CH(9) |
4.7 tot 4.8 |
1 H |
M |
|
|
CH2(10) |
5.0 |
2 H |
s |
|
|
NH(11) |
6.3 |
1 H |
m |
|
|
NH(12 of 13) |
6.98 |
1 H |
d |
Jvic = 1.5 |
|
NH(13 of 12) |
7.01 |
1 H |
d |
Jvic = 2 |
|
Ph(14 en 15) |
7.1 tot 7.3 |
10 H |
M |
|
Opmerking: De multipliciteit is niet steeds altijd duidelijk
IR:
n (in cm-1): 3330; 1740; 1670; 750 en 700
8.2.4 Synthese van cyclo[Lys(Z)-Phe] (11)[7]
In een eenhalzige 1 liter kolf wordt onder magnetisch roeren 5 g (9.23 mmol) Boc-Lys(Z)-Phe-OMe (10) opgelost in 500 ml 99% mierezuur. Men laat de oplossing gedurende 2 uur staan bij kamertemperatuur. Vervolgens evaporeert men het mierezuur door middel van een oliepomp zonder dat de temperatuur van het waterbad de 30 °C overschrijdt. Het HCOOH.H2N-Lys(Z)-Phe-OMe residu lost men op in 230 ml 2-butanol en 75 ml droge tolueen. De oplossing wordt in een 500 ml tweehalzige kolf overgeheveld. Nadat een thermometer in een hals is geplaatst, wordt de het reactiemengsel 2 uur gerefluxeert in een oliebad van 120 °C. De temperatuur in de kolf bedraagt 98 °C. Men ziet erop toe dat tijdens de reactie de hoeveelheid solvent op peil wordt gehouden en voegt eventueel 2-butanol toe. Al het solvent wordt na de 2 uur durende reactie uitgedampt en een bruto opbrengst van 16.58 g wordt verkregen. Ter zuivering wordt het residu in 400 ml hete ethylacetaat opgelost en voegt men 220 ml petroleumether toe om het diketopiperazine (11) te laten neerslaan. De neerslag filtreert men af op een glasfilter, waarna deze gewassen wordt met 200 ml petroleumether. Aldus bekomt men 3.23 g eindproduct zodat de netto opbrengst 85 % (7.89 mmol) bedraagt.
Karakterisatie:

Witte vaste stof.
Moleculair gewicht: 409.48 g/mol
Brutoformule: C23H27N3O4
Smeltdomein: 212.1 – 213.3 °C
Optische rotatie:
Solvent: CH3COOH / H2O (8 / 2)
Concentratie c = 0.0042 g/ml
[a]D= + 10°
TLC:
Eluens 1: hexaan / ethylacetaat (4 / 1)
Rf = 0
Eluens 2: isopropylether / chloroform / petroleumether (6 / 3 / 1)
Rf = 0.11
Eluens 3: ethylacetaat / methanol (4 / 1)
Rf = 0.60
De cyclo[Lys(Z)-Phe] vlek werd zichtbaar na een toluidine test.
HPLC:
|
Retentietijd/min |
Toekenning |
|
18.6 |
Cyclo[Lys(Z)-Phe] |
MS (ES+):
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
302 |
M – O-Bzl |
100 % |
|
366 |
M – CHNO |
60 % |
|
410 |
M + H |
75 % |
|
432 |
M + Na |
40 % |
|
448 |
M + K |
15 % |
NMR: (250 MHz)
Solvent: dimethyl-d6 sulfoxide
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
Koppelingsconstante J in Hz. |
|
CH(1’, 2 en 2’) |
0.7 tot 0.9 |
3 H |
M |
|
|
CH(1, 3 en 3’) |
1.0 tot 1.2 |
3 H |
M |
|
|
CH2(4’, 5 en 5’) |
2.8 tot 2.9 |
3 H |
M |
|
|
CH(4) |
3.1 |
1 H |
dd |
Jgem = 13.4 en Jvic = 3.8 |
|
CH(6) |
3.5 |
1 H |
m |
|
|
CH(7) |
4.2 |
1 H |
m |
|
|
CH2(8) |
5.0 |
2 H |
s |
|
|
Ph(9 en 10) en NH(11) |
7.0 tot 7.4 |
11 H |
M |
|
|
NH(12 of 13) |
8.0 |
1 H |
s |
|
|
NH(13 of 12) |
8.1 |
1 H |
s |
|
IR:
n (in cm-1): 3350; 3200; 1730; 1660; 755 en 700 cm-1
8.2.5 Synthese van cyclo[Lys-Phe] (12)[10]
Men lost 3 g (7.33 mmol) cyclo[Lys(Z)-Phe] (11) op in 30 ml glaciaal CH3COOH. Men giet deze oplossing in een Parr hydrogenatiefles en vervolgens suspendeert men 0.5 gewichts-equivalent (1.5 g) 10 % Pd/C in het azijnzuur. De hydrogenolyse verloopt in het Parr toestel onder een druk van 60 psi terwijl de fles voortdurend wordt geschud. Na 2 uur is de druk teruggevallen tot 52 psi en wordt de katalysator afgefiltreerd over aangestampt decaliet. Het decaliet wordt gespoeld met 50 ml CH3COOH. Men lyofiliseert het filtraat overnacht en verkrijgt een bruto opbrengst van 2.76 g. De bekomen witte vlokken lost men op in 600 ml CH2Cl2 en men voegt 220 ml petroleum ether toe teneinde het CH3COOH zout van cyclo[Lys-Phe] (12) uit te kristalliseren. Daarna filtreert men de kristallen af, men wast ze met 70 ml petroleum ether en droogt ze in een dessicator. De netto opbrengst bedraagt 2.30 g (6.86 mmol, 94 %), waarvan 4 % geepimeriseerd is. Door omkristallisatie kon de epimerisatiegraad niet gereduceerd worden.
Karakterisatie:

Witte vaste stof.
Moleculair gewicht:
cyclo[Lys-Phe]: 275.35 g/mol
CH3COOH zout: 335.40 g/mol
Brutoformule:
cyclo[Lys-Phe]: C15H21N3O2
CH3COOH zout: C17H25N3O4
Smeltdomein van het zout: 189.3 – 190.1 °C
Optische rotatie:
Solvent: 100 % CH3COOH
Concentratie c = 0.00655 g/ml
[a]D= +11 °
TLC:
Aan het in 1 ml CH2Cl2 opgeloste cyclo[Lys-Phe] (12) werd een druppel diisopropylethylamine toegevoegd vooralleer de glasplaatjes te spotten.
Eluens 1: dichloormethaan
Rf = 0
Eluens 2: acetonitril / methanol / water (4 / 1 / 1)
Rf = 0.03
Eluens 3: ethylacetaat / butanol / azijnzuur / water (2 / 1 / 1 / 1)
Rf = 0.63
De cyclo[Lys-Phe] vlek kleurde na een ninhydrine test.
HPLC:
|
Retentietijd/min |
Toekenning |
|
9.4 |
Cyclo[Lys-Phe] |
|
10.5 |
Cyclo[Lys-Phe] epimeer |
MS (ES+):
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
276 |
M + H |
100 % |
NMR: (250 MHz)
Solvent: CDCl3 met een druppel CF3COOH
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
|
CH(1’) |
0.5 |
1 H |
m |
|
CH(1, 2, 2’, 3, 3’) |
1.0 tot 1.6 |
5 H |
M |
|
CH3(4) |
2.1 |
3 H |
s |
|
CH(5’, 6, 6’) |
2.9 tot 3.1 |
3 H |
M |
|
CH(5) |
3.3 |
1 H |
m |
|
CH(7) |
3.9 |
1 H |
m |
|
CH(8) |
4.5 |
1 H |
m |
|
Ph(9) en NH3+ (10) |
6.6 tot 7.4 |
8 H |
M |
|
NH(11 of 12) |
8.0 |
1 H |
s |
|
NH(12 of 11) |
8.2 |
1 H |
s |
Opmerking: Door lijnverbreding kunnen de koppelingsconstanten niet gegeven worden.
IR:
n (in cm-1): 3200; 3100 tot 2040; 1660; 1100; 755 en 705 cm-1
8.3 Synthese van cyclo[Lys(COCH2OH)-Phe] (14)
8.3.1 Synthese van cyclo[Lys(COCH2OBzl)-Phe] (13)[11]
In een 100 ml kolf suspendeert men 100 mg (0.298 mmol) cyclo[Lys-Phe] in 25 ml CH2Cl2. Hieraan voegt men 5 ml 0.143 M NaOH (aq) toe. Men roert het bifasisch systeem dermate hevig dat de fasen niet meer van elkaar te onderscheiden zijn. Na 1.3 eq. (47 ml; 0.387 mmol) benzyloxyacetyl chloride te hebben opgelost, in 5 ml CH2Cl2voegt men deze oplossing druppelsgewijs over een periode van 10 minuten toe aan het diketopiperazine (12). Men voorziet de kolf van een stop en laat de reactie gedurende 6 u 30 doorgaan. Na een addititioneel volume van 60 ml CH2Cl2 te hebben toegevoegd, wast men de organische fase
3 keer met een 15 ml 3N HCl oplossing,
3 keer met een 15 ml 5% ige NaHCO3 oplossing,
3 keer met een 15 ml 5% ige NaCl oplossing.
Vervolgens droogt men de CH2Cl2 fase over MgSO4 en men dampt het filtraat uit. Men bekomt aldus een bruto opbrengst van 145 mg. Uit 70 ml warme ethylacetaat kristalliseert men cyclo[Lys(COCH2OBzl)-Phe] (13). Na de kristallen te hebben afgezonderd op een glasfilter, wast men ze met 15 ml petroleum ether. De opbrengst na drogen onder vacuum bedraagt 69 % (87 mg; 0.205 mmol).
Karakterisatie:

Witte vaste stof.
Moleculair gewicht: 423.51 g/mol
Bruto formule: C24H29N3O4
Smeltdomein: 192.2 – 193.5 °C
TLC:
Eluens 1: hexaan / ethylacetaat (4 / 1)
Rf = 0
Eluens 2: ethylacetaat / methanol (4 / 1)
Rf = 0.55
Eluens 3: acetonitril / methanol / water (4 / 1 / 1)
Rf = 0.87
HPLC:
|
Retentietijd/min |
Toekenning |
|
17.6 |
Cyclo[Lys(COCH2OBzl)-Phe] |
MS (ES+):
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
424 |
M + H |
100 % |
|
446 |
M + Na |
40 % |
|
462 |
M + K |
20 % |
|
847 |
2M + H |
10 % |
|
869 |
2M + Na |
20 % |
|
885 |
2M + K |
5 % |
NMR: (250 MHz)
Solvent: CDCl3 / dimethyl-d6 sulfoxide (4 / 1)
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
|
CH(1’, 2, 2’) |
0.5 tot 0.8 |
3 H |
M |
|
CH(1, 3, 3’) |
0.9 tot 1.2 |
3 H |
M |
|
CH(4, 4’, 5, 5’) |
2.1 tot 3.0 |
4 H |
M |
|
CH(6) |
3.4 |
1 H |
m |
|
CH2(7) |
3.6 |
2 H |
s |
|
CH(8) |
3.9 |
1 H |
m |
|
CH(9) |
4.3 |
2 H |
s |
|
NH(10) |
6.5 |
1 H |
m |
|
Ph(11 en 12) |
6.8 tot 7.1 |
10 H |
M |
|
NH(13 of 14) |
7.2 |
1 H |
s |
|
NH(14 of 13) |
7.3 |
1 H |
s |
IR:
n (in cm-1): 3200; 1660; 755; 700
8.3.2 Synthese van cyclo[Lys(COCH2OH)-Phe] (14)[12]
Men lost 140 mg (0.331 mmol) cyclo[Lys(COCH2OBzl)-Phe] op in een mengsel van 60 ml CH3OH en 10 ml H2O. Men voorziet de Parr hydrogenatiefles van 0.5 gewichtsequivalent (70 mg) 10 % Pd/C en men giet bovenstaande oplossing in de fles. De hydrogenolyse verloopt gedurende 5 u 30 bij een initiele druk van 48 psi. Na deze periode is de druk teruggevallen tot 46 psi. HPLC wijst uit dat nog 8 % beginproduct (13) aanwezig is. Ingevolge laat men de reactie nog 3 uur doorgaan. De katalysator wordt afgefiltreerd over aangestampt decaliet en men spoelt het decaliet met 40 ml CH3OH. Men dampt het solvent uit om een bruto opbrengst van 0.139 g te bekomen. HPLC toont aan dat enkel cyclo[Lys(COCH2OH)-Phe] (14) (Rt = 10.7) en tolueen (Rt = 23.6) aanwezig zijn. Men kristalliseert het eindproduct in 60 ml warme ethylacetaat. Nadat men de kristallen heeft afgefiltreerd, worden ze gewassen met 15 ml petroleum ether. Drogen onder vacuum levert witte kristallen met een opbrengst van 95 % (107 mg; 0.31 mol).
Karakterisatie:

Witte vaste stof.
Moleculair gewicht: 333.39 g/mol
Bruto formule: C17H23N3O4
Smeltdomein: 174.8 – 177.2 °C
TLC:
Eluens 1: dichloormethaan
Rf = 0
Eluens 2: ethylacetaat / methanol (4 / 1)
Rf = 0.25
Eluens 3: ethylacetaat / butanol / azijnzuur / water (1 / 1 / 1 / 1)
Rf = 0.76
HPLC:
|
Retentietijd/min |
Toekenning |
|
10.9 |
Cyclo[Lys(COCH2OH)-Phe] |
MS (ES+):
Solvent: methanol
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
334 |
M + H |
3 % |
|
356 |
M + Na |
100 % |
|
689 |
2M + Na |
689 |
NMR: (500 MHz)
Solvent: dimethyl-d6 sulfoxide
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
Koppelingsconstante J in Hz. |
|
CH(1’, 2, 2’) |
0.6 tot 0.8 |
3 H |
M |
|
|
CH(1, 3, 3’) |
1.0 tot 1.2 |
3 H |
M |
|
|
CH(4’) |
2.8 |
1 H |
dd |
Jgem = 13.5 en Jvic = 5 |
|
CH(5, 5’) |
2.9 |
2 H |
M |
|
|
CH(4) |
3.1 |
1 H |
dd |
Jgem = 13.5 en Jvic = 3.9 |
|
OH(6) |
3.3 |
1 H |
s |
|
|
CH(7) |
3.5 |
1 H |
m |
|
|
CH2(8) |
3.8 |
2 H |
s |
|
|
CH(9) |
4.2 |
1 H |
m |
|
|
CH(10, 10’) |
7.15 |
2 H |
d |
Jvic = 7.5 |
|
CH(11) |
7.2 |
1 H |
t |
Jvic = 7.2 |
|
CH(12, 12’) |
7.25 |
2 H |
t |
Jvic = 7.4 |
|
NH(13) |
7.5 |
1 H |
m |
|
|
NH(14) |
8.0 |
1 H |
s |
|
|
NH(15) |
8.1 |
1 H |
s |
|
IR:
n (in cm-1): 3200; 1660; 755; 700
8.4 Synthese van cyclo[Lys(CONHOH)-Phe] (4)
8.4.1 Synthese van cyclo[Lys(CONHOBzl)-Phe] (15)[15]
100 mg (0.298 mmol) cyclo[Lys-Phe] acetaat (12) wordt gesuspendeerd in 30 ml CH2Cl2. Verder gebruikt men een 100 ml kolf om 3,5 eq (0.129 g; 1.04 mmol) Na2CO3.H2O op te lossen in 20 ml H2O. Hieraan voegt men vervolgens 10 ml CH2Cl2 en 0.7 eq (0.062 g; 0.21 mmol) trifosgeen toe. Men roert de trifosgeenoplossing heftig en voegt het diketopiperazine (12) eraan toe. In een tweede 100 ml kolf voegt men 2 eq. (0.095 g; 0.596 mmol) O-benzyl-hydroxylamine hydrochloride bij 10 ml CH3OH.
Diisopropylethylamine 2 eq. (0.596 mmol; 104 ml) wordt toegevoegd om het hydroxylamine uit zijn zout te verdrijven. Na 1 uur voegt men de isocyanaat oplossing (die eruit ziet als een witte pap) bij het hydroxylamine. Dit mengsel laat men 2 uur roeren. We zonderen de CH2Cl2 fase af van de waterfase en voeren 3 extracties op de waterfase uit met 50 ml CH2Cl2. De gecombineerde CH2Cl2 fase wordt tot 80 ml uitgedampt en vervolgens wast men ze
1 keer met 25 ml H2O,
2 keer met 25 ml 1N HCl,
1 keer met 25 ml 5 % NaCl.
De organische fase wordt gedroogd over MgSO4. Na het solvent te hebben uitgedampt, bekomt men een bruto opbrengst van 60 % (75 mg). In 20 ml warme ethylacetaat kristalliseert cyclo[Lys(CONHOBzl)-Phe] (15). Nadat men de kristallen droogt onder vacuum bekomt men een opbrengst van 24 % (30 mg; 0.07 mmol).
Karakterisatie:
Witte vaste stof.
Moleculair gewicht: 424.50 g/mol
Bruto formule: C23H28N4O4
Smeltdomein: 169.7 – 171.4 °C
TLC:
Eluens 1: hexaan / ethylacetaat (4 / 1)
Rf = 0
Eluens 2: ethylacetaat / methanol (4 / 1)
Rf = 0.55
Eluens 3: ethylacetaat / methanol (4 / 1 / 1)
Rf = 0.86
HPLC:
|
Retentietijd/min |
Toekenning |
|
16.7 |
Cyclo[Lys(CONHOBzl)-Phe] |
MS (ES+):
|
m/z |
Toekenning |
Intensiteit t.o.v. de base piek (=100 %) |
|
302 |
M – NHOBzl |
40 % |
|
425 |
M + H |
100 % |
|
447 |
M + Na |
10 % |
|
849 |
2M + H |
30 % |
NMR: (250 MHz)
Solvent: dimethyl-d6 sulfoxide
|
|
Chemical shift d (in ppm) |
Integraal |
Multipliciteit |
Koppelingsconstante J in Hz. |
|
CH(1’, 2, 2’) |
0.6 tot 0.8 |
3 H |
M |
|
|
CH(1, 3, 3’) |
1.0 tot 1.2 |
3 H |
M |
|
|
CH(4’, 5, 5’) |
2.8 tot 2.9 |
3 H |
M |
|
|
CH(4) |
3.1 |
1 H |
dd |
Jgem = 13.5 en Jvic = 3.8 |
|
CH(6) |
3.5 |
1 H |
m |
|
|
CH(7) |
4.2 |
1 H |
m |
|
|
CH2(8) |
4.7 |
2 H |
s |
|
|
NH(9) |
6.5 |
1 H |
m |
|
|
Ph(10 en 11) |
7.1 tot 7.4 |
10 H |
M |
|
|
NH(12 of13) |
8.0 |
1 H |
s |
|
|
NH(13 of 12) |
8.1 |
1 H |
s |
|
|
NH(14) |
9.0 |
1 H |
s |
|
IR:
n (in cm-1): 3200; 1660; 755; 700
| home | lijst scripties | inhoud | vorige | volgende |